
[ Program Manual | User's Guide | Data Files | Databases ]
Frames shows open reading frames for the six translation frames of a DNA sequence. Frames can superimpose the pattern of rare codon choices if you provide it with a codon frequency table.
Frames plots open reading frames of a nucleic acid sequence as boxes bordered by potential start and stop codons. Potential start codons are shown as short lines that extend above the box and potential stop codons are shown as short lines that extend below the box. By default, only the start and stop codons at the ends of open reading frames are shown in the frame display; if a stop codon has been passed, no stops are shown again until a start codon is passed; if a start codon is passed, no start codons are shown again until a stop codon is passed. You can display all start and stop codons with -ALL. Frames examines all six translation frames of a sequence.
If your sequence could have intervening sequences in it, use -ALL to get Frames to show all of the possible start and stop codons in your sequence.
If you know the pattern of codon usage in the system under study, Frames marks the occurrence of rare codons in each reading frame. Use -RARe to have the rare codons marked. If you want to use a codon frequency file other than ecohigh.cod, you can specify this file with -RARe=codontablename.cod.
Here is a session using Frames to see the reading frames and mark the rare codons in the sequence Bacterial:EcoOmpa:
% frames -RARe
Plot FRAMES for what sequence? Bacterial:EcoOmpa
Begin (* 1 *) ?
End (* 2270 *) ?
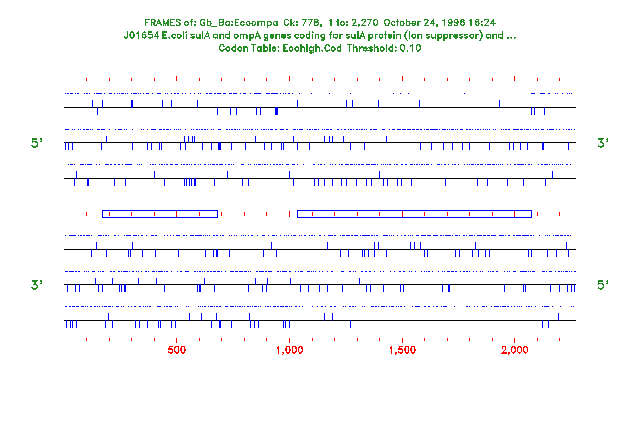
Mark rare codons at or below what threshold (* 0.0 *) ? 0.1
When your LaserWriter attached to tty07 is ready, press <Return>.
%
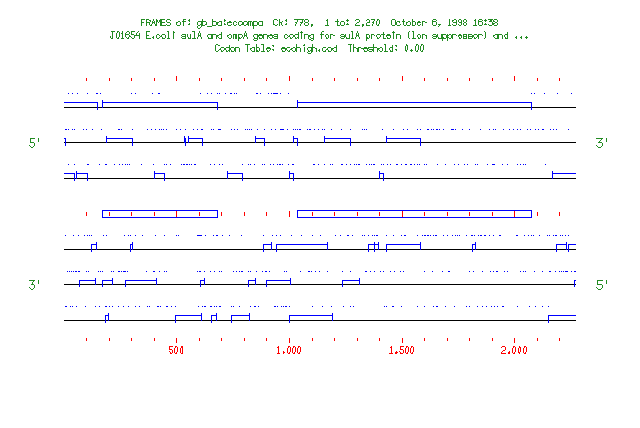
If you are reading the Program Manual, the output from this session and from another session with -ALL are shown in the two figures at the end of this program entry. In the first figure showing open reading frames, potential start codons are indicated by lines extending above the reading frame box and stop codons are indicated by lines extending below the box. In the second figure showing all potential start and stop codons, potential start codons are indicated by vertical lines above the horizontal and stop codons are indicated by vertical lines below the horizontal. In both plots, the positions of rare codons are indicated by dots above each translation frame.
Frames accepts as input a single nucleotide sequence. If Frames rejects your nucleotide sequence, see Appendix VI for information on how to change or set the type of a sequence.
Map can display the translation of open reading frames of a sequence. CodonPreference, TestCode, and Window/StatPlot help to identify coding regions. FitConsensus searches for consensus sequences.
Frames treats all the frames as open at the beginning of each strand in the range you have chosen. This could be misleading.
The Wisconsin Package must be configured for graphics before you run any program with graphics output! If the % setplot command is available in your installation, this is the easiest way to establish your graphics configuration, but you can also use commands like % postscript that correspond to the graphics languages the Wisconsin Package supports. See Chapter 5, Using Graphics in the User's Guide for more information about configuring your process for graphics.
If you need to stop this program, use <Ctrl>C to reset your terminal and session as gracefully as possible. Searches and comparisons write out the results from the part of the search that is complete when you use <Ctrl>C. The graphics device should stop plotting the current page and start plotting the next page. If the current page is the last page, plotters should put the pen away and graphic terminals should return to interactive mode.
All parameters for this program may be added to the command line. Use -CHEck to view the summary below and to specify parameters before the program executes. In the summary below, the capitalized letters in the parameter names are the letters that you must type in order to use the parameter. Square brackets ([ and ]) enclose parameter values that are optional. For more information, see "Using Program Parameters" in Chapter 3, Using Programs in the User's Guide.
Minimal Syntax: % frames [-INfile1=]Bacteria:EcoOmpa -Default Prompted Parameters: -BEGin=1 -END=2270 sets the range of interest Local Data Files: -TRANSlate=translate.txt defines start and stop codons -MARk1=ecoompa.mrk marks regions of interest on the plot Optional Parameters: -ALL shows all start and stop signals, not just open frames -RARe=ecohigh.cod marks rare codons according to codon frequency table -THReshold=0.0 sets threshold at or below which rare codons are shown -DENsity=2270 sets the number of bases per 100 platen units All GCG graphics programs accept these and other switches. See the Using Graphics chapter of the USERS GUIDE for descriptions. -FIGure[=FileName] stores plot in a file for later input to FIGURE -FONT=3 draws all text on the plot using font 3 -COLor=1 draws entire plot with pen in stall 1 -SCAle=1.2 enlarges the plot by 20 percent (zoom in) -XPAN=10.0 moves plot to the right 10 platen units (pan right) -YPAN=10.0 moves plot up 10 platen units (pan up) -PORtrait rotates plot 90 degrees
The files described below supply auxiliary data to this program. The program automatically reads them from a public data directory unless you either 1) have a data file with exactly the same name in your current working directory; or 2) name a file on the command line with an expression like -DATa1=myfile.dat. For more information see Chapter 4, Using Data Files in the User's Guide.
The translation of codons to amino acids, the identification of potential start codons and stop codons, and the mappings of one-letter to three-letter amino acid codes are all defined in a translation table in the file translate.txt. If the standard genetic code does not apply to your sequence, you can provide a modified version of this file in your working directory or name an alternative file on the command line with an expression like -TRANSlate=mycode.txt. Translation tables are discussed in more detail in Appendix VII.
If you are studying a sequence with known features, this program can mark the plot with small boxes showing the positions of these features. The presence of a file in your directory with the same name as your sequence and the filename extension .mrk causes the program to mark each range specified in the file. You can provide a marking file on the command line with an expression like -MARk=gamma.mrk. The file gamma.mrk contains information about the format of marking files. The figure for the example session shows marked regions.
You can set the parameters listed below from the command line. For more information, see "Using Program Parameters" in Chapter 3, Using Programs in the User's Guide.
Usually, translation is based on the translation table in a default or local data file called translate.txt. This parameter allows you to use a translation table in a different file. (See Appendix VII for information about translation tables.)
If you are studying a sequence with known features, this program can mark the plot with small boxes showing the positions of these features. The presence of a file in your directory with the same name as your sequence and the file name extension .mrk causes the program to mark each range specified in the file. The file gamma.mrk contains information about the format of marking files.
makes Frames show all of the potential starts and stops regardless of what precedes them in the sequence (as shown in the second figure).
If you use this parameter, Frames uses a codon frequency file of the kind written by CodonFrequency and a threshold at or below which you want to have the rare codons marked. Then, when Frames passes a codon whose value in column five of the codon frequency table falls at or below your threshold setting, the plot is marked with a dot above the codon.
sets the threshold at or below which rare codons are shown in the plot. When Frames passes a codon whose value in column five of the codon frequency table falls at or below your threshold setting, the plot is marked with a dot above the codon.
sets the number of bases or amino acids per 100 platen units (PU). This is usually equivalent to the number of bases or amino acids per page. Output from different GCG graphics programs that are run at the same density can be compared by lining up the plots on a light box. You only can set a density to compress the plot. If you specify a density that would expand the plot onto more than one page, then the density is automatically reset to plot on a single page.
The parameters below apply to all Wisconsin Package graphics programs. These and many others are described in detail in Chapter 5, Using Graphics of the User's Guide.
writes the plot as a text file of plotting instructions suitable for input to the Figure program instead of sending it to the device specified in your graphics configuration.
draws all text characters on the plot using Font 3 (see Appendix I).
draws the entire plot with the pen in stall 1.
The parameters below let you expand or reduce the plot (zoom), move it in either direction (pan), or rotate it 90 degrees (rotate).
expands the plot by 20 percent by resetting the scaling factor (normally 1.0) to 1.2 (zoom in). You can expand the axes independently with -XSCAle and -YSCAle. Numbers less than 1.0 contract the plot (zoom out).
moves the plot to the right by 30 platen units (pan right).
moves the plot up by 30 platen units (pan up).
rotates the plot 90 degrees. Usually, plots are displayed with the horizontal axis longer than the vertical (landscape). Note that plots are reduced or enlarged, depending on the platen size, to fill the page.
[ Program Manual | User's Guide | Data Files | Databases ]
Documentation Comments: doc-comments@gcg.com
Technical Support: help@gcg.com
Copyright (c) 1982, 1983, 1985, 1986, 1987, 1989, 1991, 1994, 1995, 1996, 1997, 1998 Genetics Computer Group Inc., a wholly owned subsidiary of Oxford Molecular Group, Inc. All rights reserved.
Licenses and Trademarks Wisconsin Package is a trademark of Genetics Computer Group, Inc. GCG and the GCG logo are registered trademarks of Genetics Computer Group, Inc.
All other product names mentioned in this documentation may be trademarks, and if so, are trademarks or registered trademarks of their respective holders and are used in this documentation for identification purposes only.