
[ Program Manual | User's Guide | Data Files | Databases ]
GrowTree creates a phylogenetic tree from a distance matrix created by Distances using either the UPGMA or neighbor-joining method. You can create a text or graphics output file.
GrowTree reconstructs a phylogenetic tree from a distance matrix such as the one created by Distances. Two methods are available for reconstructing the tree: UPGMA (unweighted pair group method using arithmetic averages) and neighbor-joining.
Here is a session using GrowTree to reconstruct a tree from the distance matrix created in the sample session with Distances and create both a text representation and a graphical plot of the tree.
% growtree -FIGure
What is the distance matrix ? hum_gtr.distances
Which method to use ?
1 Neighbor-joining
2 UPGMA
Choose the method to use: (* 1 *)
What should I call the trees file (* hum_gtr.trees *) ?
3 internal, 5 terminal nodes
The minimum density for a one-page plot is 3.3 taxa/100 platen units.
What density do you want (* 3.3 *) ?
That will take 1 page. Is this all right (* yes *) ?
FIGURE instructions are now being written into growtree.figure.
%
Here is the output trees file in NEXUS format:
#NEXUS
[ Trees from file: hum_gtr.distances ]
begin trees;
utree Tree_1 =
((('Gtr1_Human':18.43,'Gtr3_Human':30.18):4.34,'Gtr4_Human':24.87)
:3.19,('Gtr2_Human':35.98,'Gtr5_Human':74.88):3.19):0.00;
endblock;
Here is the output Figure file:
GrowTree accepts a distance matrix in the format produced by Distances and Diverge.
PileUp creates a multiple sequence alignment from a group of related sequences using progressive pairwise alignments. It can also plot a tree that shows the clustering relationships used to create the alignment. LineUp can be used to create and edit multiple sequence alignments. Pretty displays multiple sequence alignments and calculates a consensus sequence. It does not create the alignment; it simply displays it.
The Wisconsin Package includes several programs for evolutionary analysis of multiple sequence alignments. Distances creates a matrix of pairwise distances between the sequences in a multiple sequence alignment. Diverge measures the number of synonymous and nonsynonymous substitutions per site of two or more aligned protein coding regions and can output matrices of these values. GrowTree reconstructs a tree from a distance matrix or a matrix of synonymous or nonsynonymous substitutions. PAUPSearch reconstructs phylogenetic trees from a multiple sequence alignment using parsimony, distance, or maximum likelihood criteria; PAUPDisplay can manipulate and display the trees output by PAUPSearch and can also plot the trees output by GrowTree.
Unknown.
This method (Sneath and Sokal, Numerical Taxonomy, Freeman, San Francisco (1973)) can be used to estimate a species tree or gene tree when the expected rate of gene substitution is constant and the distance measure is linear with evolutionary time (for example, distance is measured as amino acid substitutions). The distances must be ultrametric to obtain a correct tree using this method.
The two sequences that have the smallest distance in the distance matrix are combined to form a cluster. That cluster replaces the original sequence pair as a single entry in the distance matrix (reducing the dimension of the matrix by one), and distances between the cluster and the other entries are calculated. The entries in the new matrix that have the smallest distance are combined to form a new cluster, and the process continues until only a single cluster remains. The resulting tree is a rooted tree.
Instead of using a simple average, the UPGMA method calculates the distances between a new cluster and the other entries in the distance matrix based on the total number of sequences in the cluster. If the new cluster C was formed by combining two clusters a and b, cluster a representing N(a) total sequences and cluster b representing N(b) total sequences, the distance between the new cluster C and another entry k is:
This method is designed to find an approximation to the minimum evolution tree for a set of aligned sequences, using less computer time than the full algorithm for determining a minimum evolution tree. It works best when the distances are additive. The algorithm is that of Saitou and Nei, Mol. Biol. Evol. 4; 406-425 (1987), simplified by Studier and Keppler, Mol. Biol. Evol. 5; 729-731 (1988), and modified by Swofford, Olsen, Waddell, and Hillis in Molecular Systematics, second edition, ed. Hillis, Moritz, and Mable, Sinauer Associates, Inc., 1996, Ch. 11, "Phylogenetic Inference."
The neighbor-joining method clusters the sequences in a pairwise fashion. However, instead of picking the next pair to cluster by looking for the smallest distance in the distance matrix, this method seeks to form pairs that minimize the sum of the branch lengths for the entire tree. Therefore at each round of clustering, all possible pairs of entries are considered one at a time and the sum of the branch lengths for the resulting tree is calculated. The pairing that results in the smallest sum is the one that will be used to form the new cluster. This new cluster replaces its two constituent entries in the distance matrix (reducing the dimension of the distance matrix by one), and distances are calculated between the new cluster and the remaining entries in the distance matrix. The process continues until only two entries remain. The resulting tree is an unrooted tree. Because this method attempts to build an additive tree from the data, negative branch lengths may result if the distance data are not exactly additive (see the CONSIDERATIONS topic for more information on this).
In order for these methods to produce correct trees, the steps leading up to this analysis (sequence alignment and calculation of pairwise distances) must be done carefully. For example, the alignment used to produce the distance matrix may have to be adjusted manually, especially for nucleic acid sequences that are coding regions. The proper distance correction method should be used to create the distance matrix from the alignment, depending on the characteristics of the sequences (whether the base composition or amino acid composition is skewed, whether the substitution rate varies greatly from site to site, etc.) and on the assumptions made by each of the distance correction methods.
You may notice some tree branches pointing "up" in the tree plot and negative branch lengths in the corresponding trees file. This can occur with the neighbor-joining method because the algorithm tries to represent the data by an additive tree. If the distances are not perfectly additive, negative branch lengths can result.
The appearance of negative branch lengths is not necessarily a problem. There is usually some error in calculating pairwise distances, so the distances are seldom perfectly additive. Short negative branch lengths may result from this. Short negative (or positive) branch lengths may also suggest that a polytomy exists. The neighbor-joining method reconstructs a tree by considering pairs of distances and has no method of dealing with a situation where more than two taxa branch off at the same point (a polytomy). If the branch lengths resulting from a neighbor-joining tree reconstruction are very small (either negative or positive), it may be an indication that the taxa with the short branch lengths should be represented at the same level as neighboring taxa instead of at different levels. For example, in the figure below, the tree on the right (with the trisomy) is probably a better representation than the binary tree on the left. In order to resolve a polytomy, more data will be needed.
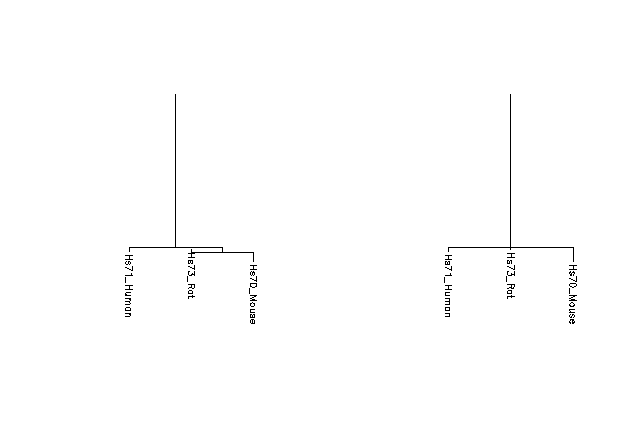
On the other hand, long negative branch lengths often indicate a problem with the distance data or with the sequence alignment from which the distance matrix was calculated. You should double-check the alignment to make sure it is the best possible alignment for the data, and examine the distance matrix used to reconstruct the tree (see the CONSIDERATIONS topic in the documentation for the Distances program). If the distance matrix contains a lot of infinite distances (represented by 999.99), the tree built from the matrix may be incorrect.
In some cases, you can create a better distance matrix. For example, if the matrix was created without correcting the distances for multiple substitutions at a single site, recreate the matrix using an appropriate correction method.
Another correction you can make is to eliminate the effects of "randomization" of the third position of codons in coding regions. Because the third position can change without altering the amino acid that the codon specifies, this position often has a much higher substitution rate than the other two positions. This contributes noise to the distance matrix. To eliminate the noise, recreate the distance matrix using only the first two positions of the codon (Distances with -POSition=4). Alternatively, translate the coding regions, align the resulting amino acid sequences, and use the protein alignment to create the distance matrix.
-NONEGative resets negative branch lengths to zero after the tree is constructed. An appropriate use of this parameter is to neaten the plot of a tree that contains negative branch lengths that are short. Do not use this parameter to disguise the fact that your tree has long negative branches! If long negative branches are present, you should be examining your data and rethinking your strategy of tree reconstruction, not worrying about visual esthetics.
When -TREEFORMat=1, a phylogram is produced and distances along the branches of the tree are indicated by a bar labeled "X substitutions per 100 residues." If the distance matrix was created by Distances, this label is correct. If the distances in your distance matrix use a metric other than substitutions per 100 residues, this label will not be correct. (In particular, distances derived from OldDistances are not expressed as substitutions per 100 amino acids.) Make sure that the label on the plot is changed to the proper units before publishing the phylogram. This can be done by running GrowTree with -FIGure=growtree.figure to create a figure file named growtree.figure, editing this file to change the label for the distance bar, and running Figure on this edited figure file to generate the tree.
All parameters for this program may be added to the command line. Use -CHEck to view the summary below and to specify parameters before the program executes. In the summary below, the capitalized letters in the parameter names are the letters that you must type in order to use the parameter. Square brackets ([ and ]) enclose parameter values that are optional. For more information, see "Using Program Parameters" in Chapter 3, Using Programs in the User's Guide.
Minimal Syntax: % growtree [-INfile=]hum_gtr.distances -Default
Prompted Parameters:
[-OUTfile=]hum_gtr.trees names the output file
(tree information in NEXUS format)
-MENu=1 uses neighbor-joining to reconstruct the tree
2 uses UPGMA method to reconstruct the tree
-DENsity=20.0 sets number of sequences/100 pu in the tree plot
Local Data Files: None
Optional Parameters:
-NONEGative resets negative branch lengths to zero
-NOBRanch suppresses reporting branch lengths in trees file
-ROUND reports branch lengths in trees file to nearest integer
-NOPLOt suppresses graphical display of tree
-ORDer=1 orders sequences in tree display using "standard" order...
=2 reverse standard order
=3 alphabetically by name (ascending)
=4 alphabetically by name (descending)
=5 "laddered" to the left by number of descendents
=6 "laddered" to the right by number of descendents
-TREEFORMat=1 draws the tree as a phylogram
=2 draws the tree as a cladogram
All GCG graphics programs accept these and other switches. See the Using
Graphics chapter of the USERS GUIDE for descriptions.
-FIGure[=FileName] stores plot in a file for later input to FIGURE
-FONT=3 draws all text on the plot using font 3
-COLor=1 draws entire plot with pen in stall 1
-SCAle=1.2 enlarges the plot by 20 percent (zoom in)
-XPAN=10.0 moves plot to the right 10 platen units (pan right)
-YPAN=10.0 moves plot up 10 platen units (pan up)
-PORtrait rotates plot 90 degrees
None.
You can set the parameters listed below from the command line. For more information, see "Using Program Parameters" in Chapter 3, Using Programs in the User's Guide.
sets the method used to reconstruct the tree: (1) neighbor joining (default), (2) UPGMA.
sets the number of sequences to plot per 100 platen units in the graphical output.
resets any negative branch lengths to 0.0 after the tree is built.
suppresses the reporting of branch lengths in the trees file.
reports branch lengths in the trees file to the nearest integer.
suppresses the output of a graphical representation of the tree.
sets the method for ordering sequences (as far as is possible) in the tree display: "standard" order (1), reverse standard order (2), order alphabetically by ascending name (3) or by descending name (4), "ladder" the taxon groups to the left (5) or to the right (6) according to the number of descendants of each internal node.
sets the format to use in drawing the tree: phylogram (1) is drawn with branch lengths proportional to calculated distances; cladogram (2) is drawn with constant branch lengths.
The parameters below apply to all Wisconsin Package graphics programs. These and many others are described in detail in Chapter 5, Using Graphics of the User's Guide.
writes the plot as a text file of plotting instructions suitable for input to the Figure program instead of sending it to the device specified in your graphics configuration.
draws all text characters on the plot using Font 3 (see Appendix I).
draws the entire plot with the pen in stall 1.
The parameters below let you expand or reduce the plot (zoom), move it in either direction (pan), or rotate it 90 degrees (rotate).
expands the plot by 20 percent by resetting the scaling factor (normally 1.0) to 1.2 (zoom in). You can expand the axes independently with -XSCAle and -YSCAle. Numbers less than 1.0 contract the plot (zoom out).
moves the plot to the right by 30 platen units (pan right).
moves the plot up by 30 platen units (pan up).
rotates the plot 90 degrees. Usually, plots are displayed with the horizontal axis longer than the vertical (landscape). Note that plots are reduced or enlarged, depending on the platen size, to fill the page.
[ Program Manual | User's Guide | Data Files | Databases ]
Documentation Comments: doc-comments@gcg.com
Technical Support: help@gcg.com
Copyright (c) 1982, 1983, 1985, 1986, 1987, 1989, 1991, 1994, 1995, 1996, 1997, 1998 Genetics Computer Group Inc., a wholly owned subsidiary of Oxford Molecular Group, Inc. All rights reserved.
Licenses and Trademarks Wisconsin Package is a trademark of Genetics Computer Group, Inc. GCG and the GCG logo are registered trademarks of Genetics Computer Group, Inc.
All other product names mentioned in this documentation may be trademarks, and if so, are trademarks or registered trademarks of their respective holders and are used in this documentation for identification purposes only.
