{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

[ Program Manual | User's Guide | Data Files | Databases ]
Overview
Types of Sequence Files
Using Database Sequences
Specifying Database Sequences by Name
Specifying Database Sequences by Accession Number
Using Single Sequence Files
Creating and Editing Single Sequences
Specifying Single Sequence Files
Specifying Sequence Type (Nucleotide or Protein)
Using List Files
Creating and Editing List Files by Hand
Programs That Create List Files
Specifying List Files
Using Rich Sequence Format (RSF) Files
Programs That Create RSF Files
Editing RSF Files
Specifying RSF Files
Using Multiple Sequence Format (MSF) Files
Programs That Create MSF Files
Editing MSF Files
Specifying MSF Sequences
Copying Database Sequence Files
Creating Sequences from Databases
Viewing Sequences
Viewing Database Sequences
Viewing Sequences in Your Directory
Reformatting Sequence Files to GCG Format
Reformatting Sequence Files
For Advanced Users
Using Personal Databases
Creating Personal Databases
Specifying Personal Databases
Refining a Sequence List
This chapter teaches you about the heart of the Wisconsin Package: using sequences. It provides information that you must know to work with sequence databases (such as GenBank, EMBL (abridged), PIR, etc.) and to use your own sequences with Wisconsin Package programs for specific analysis.
You'll learn how to
The Wisconsin Package works with many different types of sequence files:
The Wisconsin Package provides you access to nucleotide and protein database sequences. When this User's Guide was printed, the following databases were available:
To refer to sequences in these databases, use the logical names listed in the online Nucleic Acid Databases and Protein Databases tables.
Choose one of the following.
In the Nucleic Acid Databases and Protein Databases tables, you will notice that in some cases there is more than one logical name to refer to a database; use whichever you are most comfortable with. For example, to refer to sequences in GenEMBL, you could use the logical name GenEMBL or GE.
Note: Because databases are site-dependent, the
online database tables may not include all the databases available to you, or your site may name the databases
differently. In addition, because the divisions of GenBank and EMBL are subject to change, these tables may not
be complete.
To find out more about the databases, read the release notes that accompany each database release. If your site
receives the GCG Database Update Service, these release notes are located in the directory with the logical name
genmoredata. For each database, you will find a file of release notes with the name of the database and the
extension ".release". For example, to find out more about the GenBank database, type
% to genmoredata
% more genbank.release.
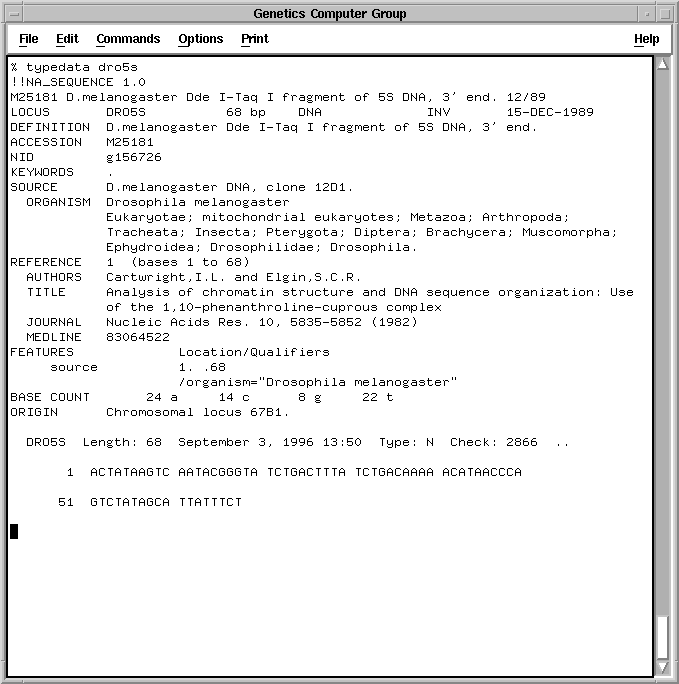
Each sequence in the databases contains not only the sequence data but also taxonomic information about the organism and the bibliographic citation. Below is an example of the sequence Dro5S from the Invertebrate division of GenBank.
You can specify database sequence entries by name. Note, however, that a sequence name is subject to change from release to release of the database. For instance, let's say an existing database sequence is merged with another sequence; the complete, merged sequence may acquire the name of the second sequence while the first sequence name is omitted. A more stable way of tracking a sequence from release to release is by its accession number, as is described in "Specifying Database Sequences by Accession Number" in this section.
Choose one of the following.
Note: Database names are case-insensitive. That is, you can type them in uppercase, lowercase, or mixed case.
There are also a number of logical names that refer to the individual divisions of GenEMBL, GenBank, EMBL, and PIR. For example, GB_In refers only to those sequences in the Invertebrate division of the GenBank database, such as GB_In:Dro5S. To refer to this same division in GenEMBL (GenBank plus EMBL), you would type Invertebrate or In, for instance In:Dro5S.
For more information on the database logical names, display the online Nucleic Acid Databases and Protein Databases tables.
The sequence names of entries in the databases sometimes change from release to release, and the same entry may have a different name in GenBank and EMBL. Because of this, publications refer to sequences by accession number. Using accession numbers offers three advantages over sequence names:
Specifying a database sequence by accession number is much like specifying one by name. Database names and accession numbers are case-insensitive. That is, you can type them in uppercase, lowercase, or mixed case.
Type the name of the database (for example, GE, which is the GenEMBL database), a colon (:), and the accession number (for example, U00069)--GE:U00069. For more information on the database logical names, see the Nucleic Acid Databases and Protein Databases tables.
Note: You cannot use wildcards to specify sequences by accession number.
If you don't know the database of the accession number, type % typedata -REF erence accession_number, for example % typedata -REFerence U00069. The program finds the sequence file in the appropriate database and displays its reference information (that is, everything but the sequence itself) on your screen. The first line of this reference information tells you the database in which the sequence resides. For example, in the illustration below, the sequence U00069 is in the Bacterial (BCT) database.
If you also want to see the sequence information, use % typedata without the -REFerence parameter. Or, if you want to copy the sequence to your directory, use the Fetch program.
When a sequence is first entered into EMBL, GenBank, PIR, or SWISS-PROT, it is assigned a unique primary accession number. If that sequence is ever merged with another sequence, the accession number of the original sequence becomes a secondary accession number in the merged sequence.
The Wisconsin Package programs treat primary and secondary accession numbers the same, as long as the accession number you use is unique. Therefore, you can access unique secondary accession numbers as well as primary accession numbers. However, if you use an accession number that occurs more than once in a database, or if you try to use an accession number that does not exist, Wisconsin Package programs will display a message saying they cannot read your sequence. If this is the case, use the LookUp program to determine the accession number's corresponding sequence name and/or primary accession number.
If the accession number you use to specify a sequence has become a secondary accession number, there is no guarantee that the sequence is exactly the same as when it had a primary accession number. That is, the original sequence may be only a portion of a new, larger entry.
You may want to find out if a primary accession number has become secondary. For example, let's say you want to view a sequence listed in a journal. However, if you retrieve that sequence by accession number from the databases, it may already have been incorporated into a larger sequence.
Choose from the following.
The reference information scrolls on your screen with the accession numbers near the top. The primary accession number always appears first, before the secondary accession numbers.
Much of the work you perform may revolve around single sequences, which are sequence files stored in your personal directories. There are three ways to create single sequence files: 1) by using SeqEd, 2) by using a text editor and the Reformat program, or 3) by using SeqLab, the graphical user interface to the Wisconsin Package.
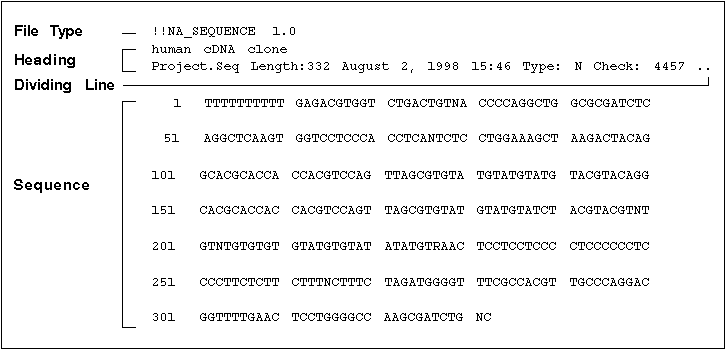
Below is an example single nucleotide sequence file created with SeqEd.
You can store single database sequences in your personal directories as well as import single sequences created by other sequence analysis software and reformat them to use with the Wisconsin Package. For more information on importing sequences, see the "Reformatting Sequence Files to GCG Format" section in this chapter.
You can create sequences from scratch in the Wisconsin Package or edit existing sequences. Each sequence must have a "type" associated with it, denoting the sequence as either a nucleotide or a protein. To specify the sequence type, you can add the parameter -NUCleotide or -PROtein to the command line when you run SeqEd or Reformat. If you forget to do so, the programs determine the type for you based on the symbols in the sequence. Note that because nucleotide and protein sequences share some symbols, the programs can guess incorrectly at the sequence type.
Choose from the following.
Heading. (optional) May contain any number of lines of text at the top of the file describing the sequence.
Dividing Line. Consists of a single line containing two periods in succession (..) to separate heading information from the sequence. This line is required only if you include heading information.
Sequence. Contains the sequence information in any format. Each line of the sequence cannot be longer than 512 characters.
Note: You also can use a text editor to modify existing sequence files, although we do not recommend this method. Once you modify a sequence with a text editor, the checksum of the sequence changes, and Wisconsin Package programs will not recognize the sequence. Therefore, if you use a text editor to modify a sequence, you must use the Reformat program to rewrite the file into GCG format.
Choose one of the following.
TIP - Sometimes the sequence files do not have characters in common; that is, you cannot use a wildcard to name several of them. If this is the case, you can create a list file to name multiple sequences. For more information, see "Using List Files" in this chapter.
Sequence type (nucleotide or protein) is an inherent part of a sequence. You can determine the type of a sequence by looking at the sequence file. Sequences in GCG format contain a dividing line between optional text heading and the sequence data. Consider the following example of a typical dividing line:
Gamma.Seq Length: 11375 August 2, 1998 10:09 Type: N Checksum: 6474 ..
The sequence type should appear on the dividing line as either Type: N for nucleotide or Type: P for protein. If the dividing line doesn't contain a Type: field, the Wisconsin Package infers the sequence type from the characters in the sequence. This inference may not always be correct.
If the Type: field of any sequence is incorrect or missing, you should correct it with the Reformat program.
Use the Reformat program. Type % reformat -NUC leotide filename or % reformat -PROtein filename. For more information, see Reformat in the Program Manual.
A list file, formerly known as a file of sequence names, is what its name implies: a file containing a list of sequence names and their locations. You can think of list files as a way to organize your sequences on a project-by-project basis.
You will find list files useful for specifying sequences from multiple files in one file that you can use as input to a program. List files can contain any number of the following types of sequences:
You can use list files with any program that accepts multiple sequences as input. A program prompt asking "What sequence(s)?" implies that the program accepts multiple sequences.
Below is an example of a list file.
In addition to sequence specifications, each sequence in a list file may optionally contain sequence attributes. These attributes include:
Begin Position. (Begin:n) Shows the base position you want to start with, where n= 1 to the length of the sequence.
End Position. (End:n) Shows the base position you want to end with, where n = 1 to the length of the sequence.
Strand. (Strand:+ or -) Defines the forward or reverse complement nucleic acid sequence strand, where + = forward strand and - = reverse strand.
Sequence Topology: Linear or Circular. (Circ:T or F) Defines the strand as linear or circular, where T = circular and F = linear.
Sequence Weight. (Wgt:n.n) Defines the sequence weight, or the significance of the sequence in comparison to other sequences. That is, you may not want all sequences accounted for equally to determine a result. Therefore, you can give some sequences greater weight than others. This attribute is of use only when you are using two or more sequences in the analysis.
Join. (Join:Sequence_Name) Indicates that the sequence segment should be concatenated with the next sequence in the list that has an identical Join: Sequence_Name attribute. Several contiguous sequences specified in a list file with the same Join:Sequence_Name attribute are concatenated together. (Assemble, Translate, and LookUp are the only Wisconsin Package programs that use the Join attribute. SeqLab uses the Join attribute to concatenate list file sequences in the Editor.)
Note: In Version 9.0 or later, the following programs use some or all of these sequence attributes in the command-line version of the Package: Assemble, CodonFrequency, Distances, Diverge, FrameSearch, PileUp, PlotSimilarity, ProfileMake, Seg, Translate, and Xnu.
File Type. (optional) Begins with the line (all uppercase) !!SEQUENCE_LIST 1.0. SeqLab uses the file type to improve performance when loading files. Do not edit or delete the file type. If present, it must appear on the first line of the file.
Description. (optional) Contains informative text, including the date of creation, describing what is in the file.
Dividing Line. (required) Includes two periods (..) that must appear on the line preceding the sequence list.
Sequence List. (required) Includes the single sequences from your personal directory or a database, sequence specifications using wildcards, RSF files, MSF files, or list files. You must provide the database or directory specification. You can add sequences in any order.
Sequence Attributes. (optional) Can include the begin and end position, indicate the forward or reverse strand, define the strand as linear or circular, give the sequence a weight in comparison with other sequences, and indicate whether the sequence is concatenated with other sequences in the list.
Sequence Comments. (optional) Includes an exclamation point (!) followed by a short comment or definition of the sequence(s) or list file.
Use a text editor of your choice and modify the file as necessary.
TIP - One way to specify a subset of sequences is to "comment out" those unwanted sequences within the list file. If you comment out sequences instead of deleting them, you can use them at a later time.
To comment out sequences:
- Open the list file in the text editor of your choice and find the sequences you do not want to use.
- Type an exclamation point (!) in front of the name of each sequence you do not want. For example
- Save the file and exit the text editor.
- To specify the list file, type an at sign (@) followed by the list filename and extension, for example @hsp70.list. The program will use only those sequences that are not commented out.
Some Wisconsin Package programs can produce output in list file format. Any program that creates multiple sequence output files and can organize those sequence specifications in a list file supports the -LIStfile parameter. You can then use that list file as input to other programs.
Programs that can create list output files and their parameters (if necessary) are listed below.
| Program | Parameter (if necessary) |
| Assemble | -LIStfile |
| BLAST | |
| Corrupt | -LIStfile |
| FastA | |
| FastX | |
| FindPatterns | -NAMes |
| FrameSearch | |
| FromEMBL | -LIStfile |
| FromFastA | -LIStfile |
| FromGenBank | -LIStfile |
| FromIG | -LIStfile |
| FromPIR | -LIStfile |
| LineUp | |
| LookUp | |
| Motifs | -NAMes |
| MotifSearch | |
| Names | |
| Pretty | -UGLy |
| ProfileSearch | |
| Reformat | -LIStfile |
| Sample | -LIStfile |
| Seg | -LIStfile |
| Simplify | -LIStfile |
| SSearch | |
| StringSearch | |
| TFastA | |
| TFastX | |
| Translate | -LIStfile |
| WordSearch | |
| Xnu | -LIStfile |
Note: Some of the programs listed above, such as LineUp and ProfileSearch, may include additional program-specific information in the output list file. Others, such as FastA and BLAST, may include sequence alignments. This extra information does not affect the list file's performance.
Type an at sign (@) and the name of the list file and extension, for example @hsp70.list.
Note: You cannot use wildcards to specify a list file. For example, you cannot specify @hsp*.list.
A Rich Sequence Format (RSF) file contains one or more sequences that may or may not be related. In addition to the sequence data, each sequence can be richly annotated with descriptive sequence information such as:
RSF files are powerful for using with SeqLab, the graphical user interface to the Wisconsin Package. Because they store positional information, you can display RSF files within SeqLab's Editor mode to view and edit sequence alignments and features. The features annotation allows you to graphically view and align sequences based on features as well as run programs on sequence regions selected by feature. You also will find RSF files useful for distributing sequences to colleagues, since these files contain each sequence's data and descriptive information.
Note: If you plan on using SeqLab for the bulk of your analyses, it is best to save your files as RSF if possible. RSF files are more richly annotated than list files or MSF files, which do not save sequence features information as part of the file.
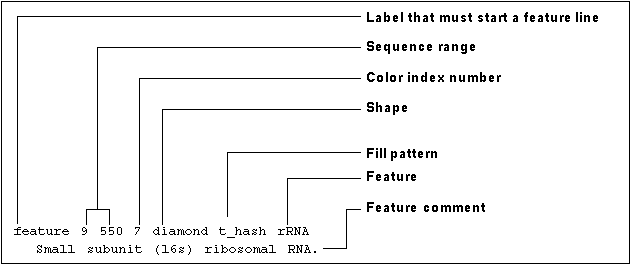
Below is an example of an RSF file.
You may find the following components in an RSF file:
The colors, shapes, and fill patterns depicted in SeqLab's Editor are defined in a resource file called feature.cols. To customize these attributes, copy feature.cols to your current directory by typing % fetch feature.cols. Then edit the file in the text editor of your choice, for example vi. The file is internally documented.
Choose from the following.
| Program | Parameter (if necessary) |
| CoilScan | -RSF |
| FindPatterns | -RSF |
| FrameSearch | -RSF |
| HTHScan | -RSF |
| Map | -RSF |
| Motifs | -RSF |
| MotifSearch | -RSF |
| NetFetch | |
| PeptideMap | -RSF |
| PeptideStructure | -RSF |
| Prime | -RSF |
| Reformat | -RSF |
| SPScan | -RSF |
Use SeqLab. If you load an RSF file into SeqLab's Editor, it graphically displays the sequences in the file. For more information, see Chapter 2, Editing Sequences and Alignments in the SeqLab Guide.
You can also use a text editor to modify an RSF file. If you do, however, the file's checksum changes, and Wisconsin Package programs will not recognize the file. Therefore, if you use a text editor to modify an RSF file, you must use the Reformat program with the -RSF parameter to rewrite the file into GCG format.
Choose one of the following.
You can combine multiple sequences in a single file, called a Multiple Sequence Format (MSF) file. MSF files include not only the sequence name but also the sequence itself, which is usually aligned with the other sequences in the file. You can specify a single sequence within an MSF file, a subset of sequences, or all sequences. Like other sequences, those in an MSF file can be used with other Wisconsin Package programs.
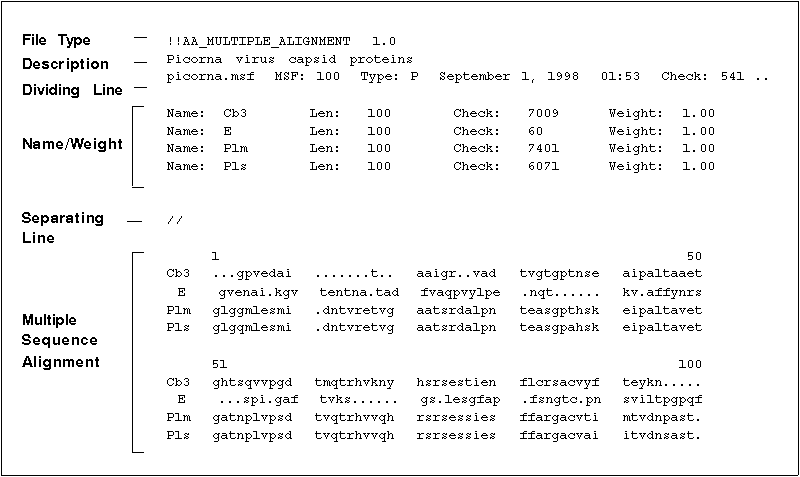
The following illustration shows an MSF file created with PileUp.
You may find the following components in an MSF file:
Choose from the following.
| Program | Parameter (if necessary) |
| LineUp | -MSF |
| PileUp | |
| PrettyBox | |
| ProfileGap | -MSF |
| ProfileSegments | -MSF |
| Reformat | -MSF |
Note: If you use % reformat -MSF to create an MSF file, it does not align the sequences.
Use LineUp. For more information, see LineUp in the Program Manual.
You also can use a text editor to modify an MSF file. If you do so, however, the file's checksum changes, and Wisconsin Package programs will not recognize the file. Therefore, if you use a text editor to modify an MSF file, you must use the Reformat program with the -MSF parameter to rewrite it into GCG format.
Choose from the following.
Note: You cannot use wildcards to name an MSF filename (that is, you cannot specify pic*.msf). You can use wildcards only between the curly brackets { }. Also, an MSF sequence specification must contain a sequence name or wildcard within the curly brackets. The MSF filename alone is not enough.
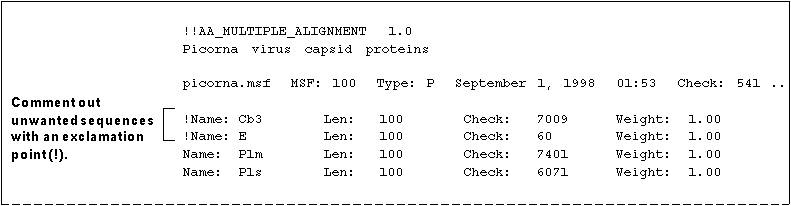
TIP - One way to specify a subset of sequences is to "comment out" those unwanted sequences within the MSF file. If you comment out sequences instead of deleting them, you can use them at a later time.
To comment out sequences:
- Open the MSF file in the text editor of your choice and find the sequences you do not want to use in the Name/Weight area toward the top of the file.
- Type an exclamation point (!) in front of the "Name:" of each sequence you do not want. For example
- Save the file and exit the text editor.
- In response to a program prompt, type the MSF filename and extension followed by an asterisk (*) wildcard in curly brackets, for example picorna.msf{*}. The program will use only those sequences which are not commented out.
The Wisconsin Package makes it easy for you to copy sequences from databases to your directory. You can copy single or multiple sequences from your local databases using Fetch or from NCBI using NetFetch. For additional information on Fetch and NetFetch, see the Program Manual.
Choose from the following.
TIP - If you know the database in which a sequence resides, you can speed its retrieval by including the database in the entry name specification, for example % fetch In:Dro5S.
To copy a single sequence from NCBI, type % netfetch entry_name or % netfetch accession_number, for example % netfetchz z12136. The sequence is retrieved and stored in an RSF file in your current directory.
TIP - You also can copy multiple sequences from the databases by creating a list file of those sequences of interest (see "Using List Files" in this chapter for more information). This method is useful if the sequence names do not have characters in common. Then, to copy the sequences from the database, type % fetch @ list_filename, for example % fetch @hiv-gag.list. The sequences in the list file are copied to your current directory as separate sequences.
To copy multiple sequences from NCBI, indicate the name of a NetBLAST output file, for example % netfetch zea2_maize.blastp. The sequences are retrieved and stored in an RSF file in your current directory.
You may want to read the reference information associated with a sequence or view the sequence itself. You can easily view the contents of sequence files by using the TypeData program. Using these commands, you can view database sequences or those in your personal directories, including single sequences, RSF files, MSF files, or list files.You can easily view the contents of sequence files by using the TypeData program. Using this command, you can view database sequences or those in your personal directories, including single sequences, RSF files, MSF files, or list files.
Note: You can also use SeqLab, the graphical interface to the Package to view and edit sequences. For more information, see the SeqLab Guide.
Type % typedata entry_name , for example % typedata GB_IN:Dro5S. The sequence data, including reference information, scrolls on your screen. Note that you cannot edit a file using the TypeData command.
You can control screen output in the following ways:
For more information on controlling screen output, see "Controlling Screen Output" in the "Quick Reference" section of Chapter 1, Getting Started.
Type % more filename, for example % more gamma.seq. The sequence data, including reference information, displays one screen at a time. To advance from screen to screen, press the <Space Bar>.
At some point in your work with the Wisconsin Package, you may need to reformat sequence files into GCG format. This may happen when
You can use a number of differently formatted sequences with the Wisconsin Package--sequences created with a text editor or automated sequencer; sequences in a different software format (for example Staden or IntelliGenetics); or sequences in the database formats of GenBank, EMBL, PIR, or SWISS-PROT.
Each sequence in the Wisconsin Package must have a "type" associated with it, denoting the sequence as either a nucleotide or a protein. To specify the sequence type, you can add the parameter -NUCleotide or -PROtein to the command line when you run Reformat, FromStaden, FromEMBL, FromFastA, FromGenBank, FromPIR, or FromIG. If you forget to do so, the programs will determine the type for you based on the symbols in the sequence. Note that because nucleotide and protein sequences share some symbols, the programs can guess incorrectly at the sequence type.
Choose one of the following.
Note: If the sequence file contains descriptive or reference information in addition to the sequence information, you first must open the file in a text editor and insert a line that contains two periods (..) above the sequence information. Then use Reformat to rewrite the sequence to GCG format.
Note: You can use Staden sequences directly with the Wisconsin Package without reformatting them by adding -STAden to the command line when you run a Wisconsin Package program.
Note: You can use FastA sequences directly with the Wisconsin Package without reformatting them by adding -FASTA to the command line when you run a Wisconsin Package program.
The information in this section is intended for users who are familiar with using sequences within the Wisconsin Package. This section teaches you how to
You can create your own personal databases, similar to GenBank and EMBL databases, for searching with the Wisconsin Package. This option is a particular advantage if you frequently work with large list files. A large set of sequences is more compact to store and faster to search if it is assembled into a database. Thus, you can convert your large list files into databases for faster searching capabilities. When sequences are assembled into a database, all Wisconsin Package programs work with them exactly as they work with public databases (GenBank, EMBL, PIR, etc.).
The program DataSet creates databases from any set of sequences you specify.
The program displays the prompt "What should I call the database?"
Your personal database logical names are automatically assigned in a shell script called .datasetrc in your home directory.
Specifying a personal database you created using DataSet is the same as specifying a sequence from a public database such as GenEMBL, GenBank, PIR, etc.
Type the logical name of your database, followed by a colon (:), followed by the sequence(s) of interest. For instance, using the example above, you could type HSP:Hs70_Brelc to specify a single sequence in the personal database, or HSP:* to specify all sequences in the personal database. For more information, see "Using Database Sequences" in this chapter.
You can refine list files, RSF files, or MSF files to fit your analysis needs:
For more information on the above programs, see the Program Manual.
Note: You cannot combine MSF files in this way.
Note: You cannot "comment out" sequences in RSF files in this way.
[ Program Manual | User's Guide | Data Files | Databases ]
Documentation Comments: doc-comments@gcg.com
Technical Support: help@gcg.com
Copyright (c) 1982, 1983, 1985, 1986, 1987, 1989, 1991, 1994, 1995, 1996, 1997, 1998 Genetics Computer Group Inc., a wholly owned subsidiary of Oxford Molecular Group, Inc. All rights reserved.
Licenses and Trademarks Wisconsin Package is a trademark of Genetics Computer Group, Inc. GCG and the GCG logo are registered trademarks of Genetics Computer Group, Inc.
All other product names mentioned in this documentation may be trademarks, and if so, are trademarks or registered trademarks of their respective holders and are used in this documentation for identification purposes only.