Transcriptomic Deconvolution of Heterogeneous Human Tissues
02/06/2024
data.RmdBackground
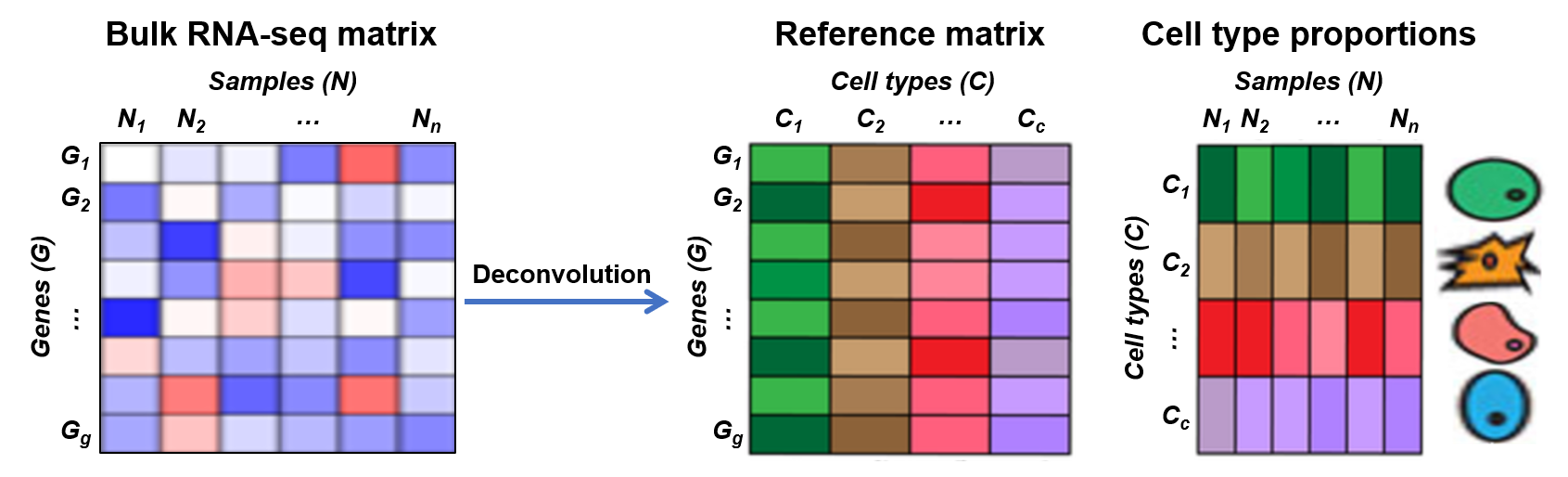
Transcriptome deconvolution of bulk tissues refers to estimating the cellular proportions in the tissue samples based on the gene expression data. It is a commonly used technique for understanding the heterogeneity of the tumor or disease tissues and further the microenvironment to facilitate downstream functional analysis. Single-cell-based deconvolution methods employ single-cell profiling data as references to deconvolve bulk tissues, which forms a multivariate regression problem.
Dataset Description
This dataset was obtained from 7 high-grade serous ovarian (HGSO) tumor samples collected by the Penn Ovarian Cancer Research Center. Each tumor was subjected to comprehensive profiling through four distinct technologies, yielding four types of sequencing data: three types of bulk RNA-seq data (polyA+ dissociated, rRNA-dissociated, and rRNA-Chunk) and the matched single-cell RNA-seq (scRNA-seq) data. The scRNA-seq data was used to construct the pseudo-bulk RNA-seq data, calculate the true cell type proportions, and obtain a reference matrix for deconvolving the bulk RNA-seq samples.
The bulk RNA-seq data is organized into matrices, one for each type of RNA-seq. For each matrix, rows represent genes, and columns correspond to samples. In other words, the (i,j)-th entry of each matrix denotes the expression of the i-th gene in the j-th bulk sample. The total number of genes is 17,109. Each of the seven HGSO samples has three replicates, resulting in a total of nine 17109-by-7 matrices.
Same as the bulk RNA-seq data, the pseudo-bulk RNA-seq data is organized into matrices as well, with rows representing genes and columns representing samples. it is obtained from the scRNA-seq data by summing up the gene expressions across all cells within each scrna-seq sample. therefore, we have three 17109-by-7 matrices, one for each replicate.
The true cell-type proportions are also organized into a matrix, whose rows representing cell types (13 in total) and columns representing samples (7 in total). The 13 different cell types include 5 different lymphcytes (B cells, plasma cells, T cells, natural killer (NK) cells, and innate lymphatic cells (ILCs)), 5 different myeloid cells (monocytes, dendritic cells (DCs), plasma DCs (pDCs), macrophages, and mast cells), endothelial cells, fibroblasts, and epithelial cells. Note that not all 13 cell types are present in each of the 7 samples. For example, Sample-2283 lacks B cells.
The reference matrix obtained from the scRNA-seq data is the key information used for estimating the cell type proportions. It denotes the average gene expression at the cell type level. Specifically, the (i,j)-th entry of the reference matrix denotes the average expression of the i-th gene in the j-th cell type. Reference matrices are sample specific, which means one reference matrix is calculated from one sample and then used to deconvolve the bulk data of that specific sample. In practice, however, people prefer using the consensus reference matrix for bulk deconvolution. The consensus reference matrix is obtained by averaging all sample specific reference matrices. It ensures a comprehensive representation of cell types since certain cell types may be absent in the sample specific reference matrices. The sample specific reference matrices are valuable for assessing the robustness of deconvolution methods against various confounding factors, including the absence of certain cell types and sample specific variations. We provide the seven sample specific reference matrices and the consensus reference matrix in this data set.
To facilitate exploration of single-cell data and assessment of deconvolution frameworks, we offer seven annotated Seurat objects derived from scRNA-seq data.
Cell type annotations are available in the ‘annotation’ column in each Seurat object. For detailed analysis of scRNA-seq data using Seurat objects,
refer to the official tutorial at
https://satijalab.org/seurat/articles/pbmc3k_tutorial.
Challenges
1)Technological discrepancies. Technological discrepancies between bulk and single-cell techniques during sample acquisition, preparation, and sequencing can lead to substantial gene expression inconsistencies. These discrepancies form a main concern hindering the accurate estimation of cell type propositions. Identifying and down-weighting genes impacted with high technological discrepancies could enhance the accuracy, as shown in a recent study (Guo et al. 2023).
2)Missing cell types. The deconvolution literature assumes that the collected samples are representative and contains all the cell types from the population. However, the sample preparation process could cause missing of certain specific cell types. That means, all samples uniformly miss certain cell types. This challenge limits the estimation accuracy of existing single-cell based deconvolution methods and necessitates the development of new methods that can robustly handle incomplete cell type profiles.
3)Inter-tumor heterogeneity. Inter-tumor heterogeneity presents a significant challenge for deconvolution analysis by introducing variability in cell type compositions and expression profiles across different tumor samples. Finding and using genes that are biologically stable across different samples to deconvolve tumor bulk samples might provide a solution to address the inter-tumor heterogeneity.
Reference
[1] Guo, Shuai, Xiaoqian Liu, Xuesen Cheng, Yujie Jiang, Shuangxi Ji, Qingnan Liang, Andrew Koval, et al. 2023. "DeMixSC: A Deconvolution Framework That Uses Single-Cell Sequencing Plus a Small Benchmark Dataset for Improved Analysis of Cell-Type Ratios in Complex Tissue Samples." bioRxiv.
[2] Hippen, Ariel A, Dalia K Omran, Lukas M Weber, Euihye Jung, Ronny Drapkin, Jennifer A Doherty, Stephanie C Hicks, and Casey S Greene. 2023. "Performance of Computational Algorithms to Deconvolve Heterogeneous Bulk Ovarian Tumor Tissue Depends on Experimental Factors." Genome Biology 24 (1): 239.