Statistical Bioinformatics Lab
Cancer is a complex disease shaped by layers of genetic and transcriptomic heterogeneity. The Wang laboratory is dedicated to advancing statistical bioinformatics to unravel this complexity. We develop computational frameworks that uncover the dynamics of tumor evolution and tumor microenvironment (i.e., cell-type specific transcriptional activities), and clonal architecture across diverse cancer types. We developed tools such as MuSE to enable fast and accurate mutation calling, or DeMixT to provide robust tumor-specific transcriptome deconvolution. Additionally, with over 15 years of experience in cancer risk modeling, we utilize Bayesian statistics and machine learning to develop software tools for clinical cancer prevention and prognosis. Our current research directions are 1) multi-omic deconvolution to study DNA–RNA dynamics in cancer, and 2) cancer risk modeling using machine learning and Bayesian models. In collaboration with clinicians and experimental biologists, we translate these insights into testable hypotheses and clinically meaningful advances. We are equally committed to fostering a research community where statistical rigor and artificial intelligence come together to push the boundaries of cancer discovery.
Pre-doctoral and post-doctoral fellow positions are available (see the cancer genomics position). Please inquire with Dr. Wang.


Current Research Directions
Multi-omic deconvolution to study DNA–RNA dynamics in cancer
Cancer is driven by genetic mutations, including single nucleotide variations (SNV), copy
number alterations (CNA), and structural variations (SV), which influence tumor behavior,
such as growth rate, treatment resistance, and metastasis. Identifying these mutations is
critical for cancer research. While whole-genome sequencing (WGS) and whole-exome sequencing
(WES) are key tools, basic steps like somatic mutation calling can be slow, limiting
large-scale analysis. My lab is addressing this with [MuSE2], a fast and efficient
mutation
calling method that facilitates large dataset analysis and advances precision
medicine. We
are also interested in improving methods for reconstructing subclonal structures,
which are
critical for understanding cancer evolution and treatment resistance. Our effort on
developing software tools like [CliPP] helps overcome
limitations in previous methods by
significantly reducing computational resources and time, through penalized likelihoods [Characterizing
ITH]. These advancements are critical to understanding intratumor
heterogeneity and cancer evolution, providing important evidence for translational research
to improve patient outcomes.
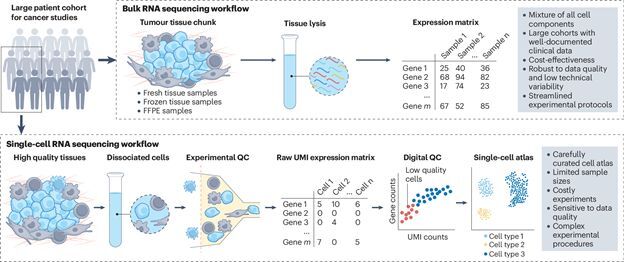
Tissues, including tumors, contain diverse cell types, each with unique transcriptional
patterns that can be studied through RNA expression data. While Single-cell RNA
sequencing (scRNA-seq) provides detailed insights, it is often costly and
challenging for large-scale
use. Bulk RNA-seq is more affordable but mixes signals from different cell types. To
address
this, deconvolution methods like [DeMixSC] help separate
these signals, improving analysis
of cell proportions and disease mechanisms. In cancer research, deconvolution differentiates
tumor from non-tumor cells, offering insights into pathways, prognosis, and heterogeneity
[DeMixT]. We further
developed an
integrative transcriptomic/genomic deconvolution method to
calculate [TmS]
(tumor-specific total mRNA expression), a feature of cancer cell plasticity,
with a striking ability to predict prognosis across cancers. Spatial transcriptomics
data builds on this by adding another dimension, preserving the spatial arrangement of cells
to
help
map tumor microenvironments (TME). This spatial context provides crucial insights into how
cells
interact within their environments, which is essential for understanding tumor progression.
We recently developed DeMixNB to characterize spatial distributions of tumor-specific
gene
expression. By integrating bulk, single-cell, and spatial data, we can achieve deeper
insights, advancing more effective and personalized cancer treatment strategies.
Cancer Risk Modeling (TP53) using machine learning and Bayesian models
Cancer survivors represent a fast-growing yet under-studied population with respect to cancer
risk, particularly for second primary cancers, which frequently occur in survivors of breast
and bladder cancer. Current risk assessments often overlook prior cancers due to limitations
in large databases like SEER, which mainly account for age and sex. To address this, my lab
studies patients with Li-Fraumeni syndrome (LFS), a hereditary condition
linked
to higher cancer risk. LFS patients often develop multiple primary cancers, offering a
unique opportunity to study cancer risk while accounting for additional factors like
mutation status. Using LFS data, we developed [LFSPRO] to predict both
first and second primary tumors in LFS families. These insights can help physicians and
genetic counselors provide personalized treatment and screening plans, aiming for early
detection of cancers in survivors and LFS patients' Personalized Risk Prediction.
We are also particularly interested in the biological annotation of TP53 mutations,
as the
germline mutations of TP53 are the main cause of LFS. Known as the “guardian of the
genome”,
the TP53 gene plays a critical role in cell signaling, apoptosis, metabolism, DNA
repair and
transcription, and in the meantime it is the most frequently mutated gene in human cancer.
We developed Survival-based clustering of predictors [SCP] using penalized likelihoods
for
survival outcomes, to cluster hundreds of TP53 missense mutations in terms of their
associated early, medium and late onset of cancer in LFS. This research aims to uncover new
patterns in cancer susceptibility and improve predictive models, offering deeper insights
into the genetic underpinnings of cancer risk in LFS patients.
PI: Wenyi Wang
Department of Bioinformatics and Computational Biology
Wenyi Wang (王文漪), Professor, Department of Bioinformatics and Computational Biology, Division of Basic Science Research, The University of Texas MD Anderson, Cancer Center, Houston, Texas
Curriculum Vitae
News Highlights

Congratulations to Ruonan Li on her publication in The American Journal of Human Genetics!
We are thrilled to share that Ruonan's paper, "Performance of LFSPRO prediction in TP53 mutation
status for prospectively collected probands," has been accepted by The American Journal of Human Genetics.
In this study, we prospectively evaluated LFSPRO in a real-world clinical genetic counseling setting, demonstrating its substantially higher accuracy than current guidelines and strong concordance with genetic counselors' clinical judgment.
Free access at
https://doi.org/10.1016/j.ajhg.2026.03.014.

Congratulations to Yaoyi Dai on her publication at Cell Reports Medicine today!
We are thrilled to share that Yaoyi's paper, "Tumor Microenvironment Transcriptional Activity Enables Robust Stratification of Chemotherapy Response in Triple-Negative Breast Cancer," has been published by Cell Reports Medicine.
In this study, we introduced tumor-specific total mRNA expression (TmS), a novel metric derived from matched RNA/DNA sequencing to robustly stratify triple-negative breast cancer. TmS outperforms established subtypes in predicting chemotherapy outcomes across diverse populations, identifying conserved stromal features and therapeutic vulnerabilities in treatment-resistant tumors. See press release from MD Anderson. .

Jessica joins UT Arlington as Tenure-Track Assistant Professor!
Congratulations to Jessica on her new role as a tenure-track Assistant Professor at UT Arlington, Cook Children's Hospital. Thank you for your dedication and impact in the lab! We wish her the very best in this exciting new career step.
"A Guide to Transcriptomic Deconvolution in Cancer" is out on Nature Reviews Cancer!
Our review on transcriptomic deconvolution in cancer has been published in Nature Reviews Cancer. It covers 43 methods, provides a practical framework for choosing among them, and highlights applications ranging from tumor microenvironment profiling to treatment response prediction. Read our interview with StatsUpAI and the review paper -> click here:

Congratulations to Graduate Student Hao Yan!
Hao has joined Biogen as a Spatial Transcriptomics Co-op in the Computational Biology group within the Biomarkers and Systems Biology department. We're excited for you and look forward to hearing about your work!.

Wang Lab Celebrates 15th Anniversary with Retreat at Enchanted Rock
Members of the Wang Lab gathered at Enchanted Rock State Natural Area, Texas, for a hiking retreat to celebrate the lab's 15th anniversary. The outing provided an opportunity to reflect on years of collaboration and scientific progress while enjoying the Texas Hill Country landscape. The retreat highlighted the strong sense of community within the lab and marked an important milestone in its continued growth and success.

Congratulations to Wenyi and Judy!
Congratulations to Professors Wenyi Wang and Huixia Judy Wang (Rice University) for receiving a Cancer Bioengineering Collaborative Seed Grant to advance conformal inference methods for predicting immunotherapy benefit.

We are now recruiting postbaccs!
The Postbacc-QB program at MD Anderson Cancer Center is now accepting applications for its next cohort of trainees interested in quantitative biology and cancer research. The program is directed by Dr. Wenyi Wang, who leads efforts to mentor and develop future scientists through immersive, hands-on research training.
Learn more and apply here: https://odin.mdacc.tmc.edu/~wwang7/Postbacc.html
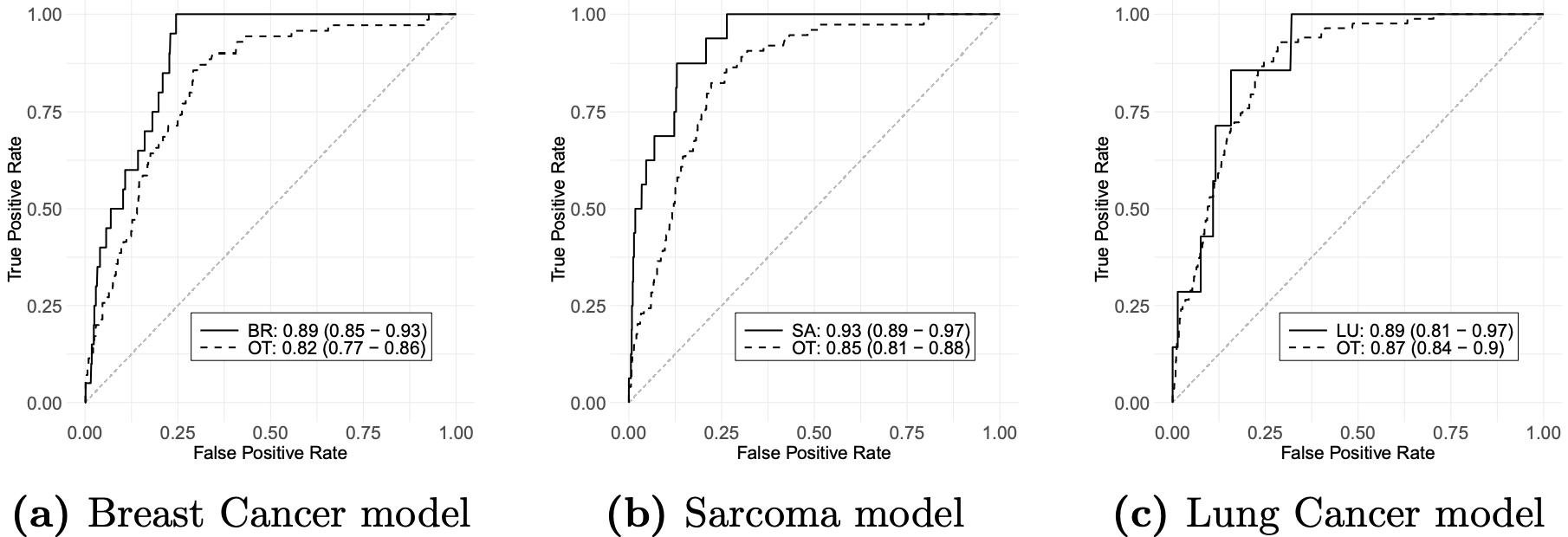
Personalized Risk Prediction for Cancer Survivors: A Bayesian Semi-parametric Recurrent Event Model with Competing Outcomes has been accepted at Annals of Applied Statistics (AOAS)
This work introduces a Bayesian semi-parametric recurrent event model with competing outcomes, enabling covariate-adjusted age-to-onset penetrance curves and delivering strong predictive performance for second primary cancers (AUC: Lung ≈ 0.89, Sarcoma ≈ 0.91, Breast ≈ 0.76).
These findings move us closer to personalized healthcare for cancer survivors by refining risk prediction and supporting clinical decision-making.
Preprint available: https://www.biorxiv.org/10.1101/2023.02.28.530537v2

Congratulations to Dr. Montierth!
We are delighted to congratulate Matthew Montierth on the successful defense of his PhD dissertation and the completion of his doctoral journey. May this achievement be the beginning of many more accomplishments ahead!

Congratulations to Hao Yan!
Our PhD student Hao Yan has been awarded the Dr. M. Stewart West Memorial Scholarship in Biometry for 2025–2026. We look forward to his continued growth and accomplishments in his academic journey!

Congratulations to Dr. Guo!
Congratulations to Shuai Guo for successfully defending his PhD thesis and earning his doctorate! Wishing him all the best in his future endeavors!

Xiaoqian's transfer learning study on TP53 Wins IMS FSML Travel Award!
We’re excited to announce that our work, "Transfer Learning for Survival-based Clustering of Predictors with an Application to TP53 Mutation Annotation," has won the IMS Frontiers of Statistical Machine Learning (FSML) Travel Award after a competitive review process. This award recognizes our innovative approach in combining transfer learning and survival analysis, with potential applications in related cancer research. We look forward to presenting our work at the FSML workshop in August 2025.

Wang Lab at AACR 2025
This April, the Wang Lab participated in the AACR 2025 conference, where we had the opportunity to present 6 posters showcasing our recent research. Congratulations to lab member undergraduate researcher Aaron Wu’s poster on CliPP-on-Web being featured by MD Anderson’s LinkedIn page.

Dr. Wenyi Wang to present at RECOMB-CCB 2025 as a keynote speaker
Wenyi was invited to present the groundbreaking work from our latest research at RECOMB-CCB 2025, a leading conference in computational cancer biology. Her keynote, 'Deciphering Tumor Heterogeneity for Benefits from Immunotherapy in Cancer,' provided new perspectives and contributed to ongoing discussions in the field.

Our immune profiling paper is published in the prestigious JASA!
Our collaborative work, "Immune Profiling Among Colorectal Cancer Subtypes
Using Dependent Mixture Models,"
is now published in the Journal of the American Statistical
Association.
In this study, we developed a novel Bayesian modeling approach to compare T
cell subtypes between early- and late-onset
colorectal cancer. The model identifies immune cell populations that are
condition-specific or shared, helping uncover
mechanisms linked to tumor progression and potential treatment strategies.
Congratulations to Yunshan and Shuai!

DeMixSC paper is published at Genome Research!
DeMixSC, an innovative deconvolution approach that overcomes the technological discrepancy between bulk and sc/snRNA-seq data using an improved wNNLS framework. It achieves high accuracy and generalizability, requiring only a small tissue-matched benchmark dataset for the targeted large bulk cohorts.
Free access at https://genome.cshlp.org/content/35/1/147
Exciting Updates from Stats Up AI
We’re thrilled to share two major milestones for Stats Up AI:
1.ASA Approval:
The Stats Up AI interest group has been officially approved by the
American Statistical Association, recognizing its mission to foster
collaboration and innovation at the intersection of statistics and AI.
2.Harvard Data Science Review Publication:
Our recent event, Stats and AI- A Fireside Conversation, was a great
success. A summary of the discussion has been accepted for publication
in the Harvard Data Science Review, further amplifying its impact.
These achievements highlight the growing influence of Stats Up AI in
advancing
the integration of statistics and AI. Stay tuned for future updates as we
continue to engage with this inspiring community!

Congratulations to Carissa for winning the ABRCMS Presentation Award!
Congratulations to our summer intern, Carissa Fong, for winning the presentation award at ABRCMS (Annual Biomedical Research Conference for Minoritized Scientists)! Her award-winning presentation showcased machine learning approaches to effectively predict tumor-specific mRNA expression (TmS). We are so proud of her achievement!

Wang Lab Postdoc Ankita Paul Awarded MD Anderson IDSO Fellowship
Congratulations to our postdoc Ankita, who has been awarded the MD Anderson Institute for Data Science in Oncology (IDSO) Fellowship! This fellowship is a great opportunity that provides junior researchers with advanced training in applying data science to oncology. We are excited to see the impactful contributions she will make through this program!
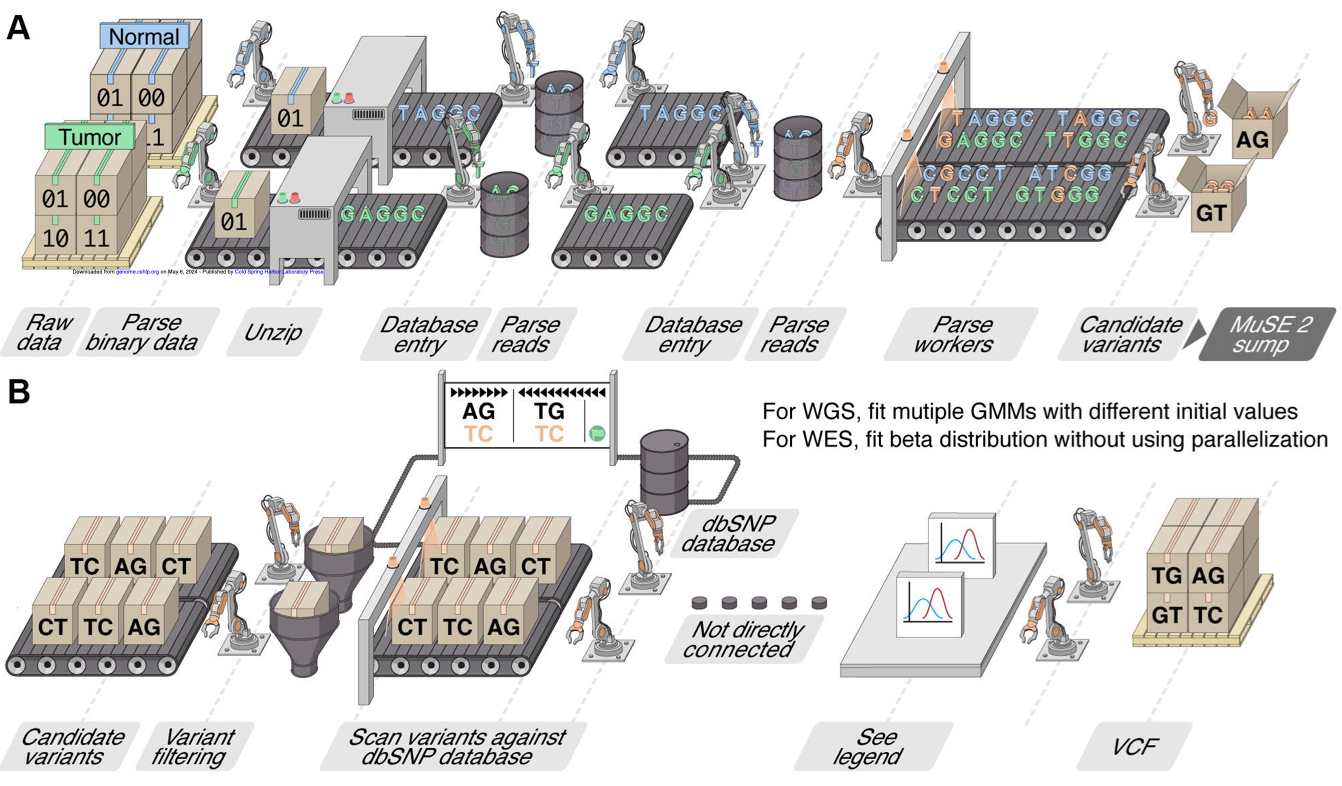
MuSE2.0 paper is online at Genome Research online!
We are exctied to officially introduce MuSE2.0, which reduces computing time by up to 50x compared to MuSE 1.0 and 8-80x compared to other popular callers. Our benchmark study suggests combining MuSE2.0 and the recently accelerated Strelka2 achieves high efficiency and accuracy in analyzing large cancer genomic datasets.
Free access here
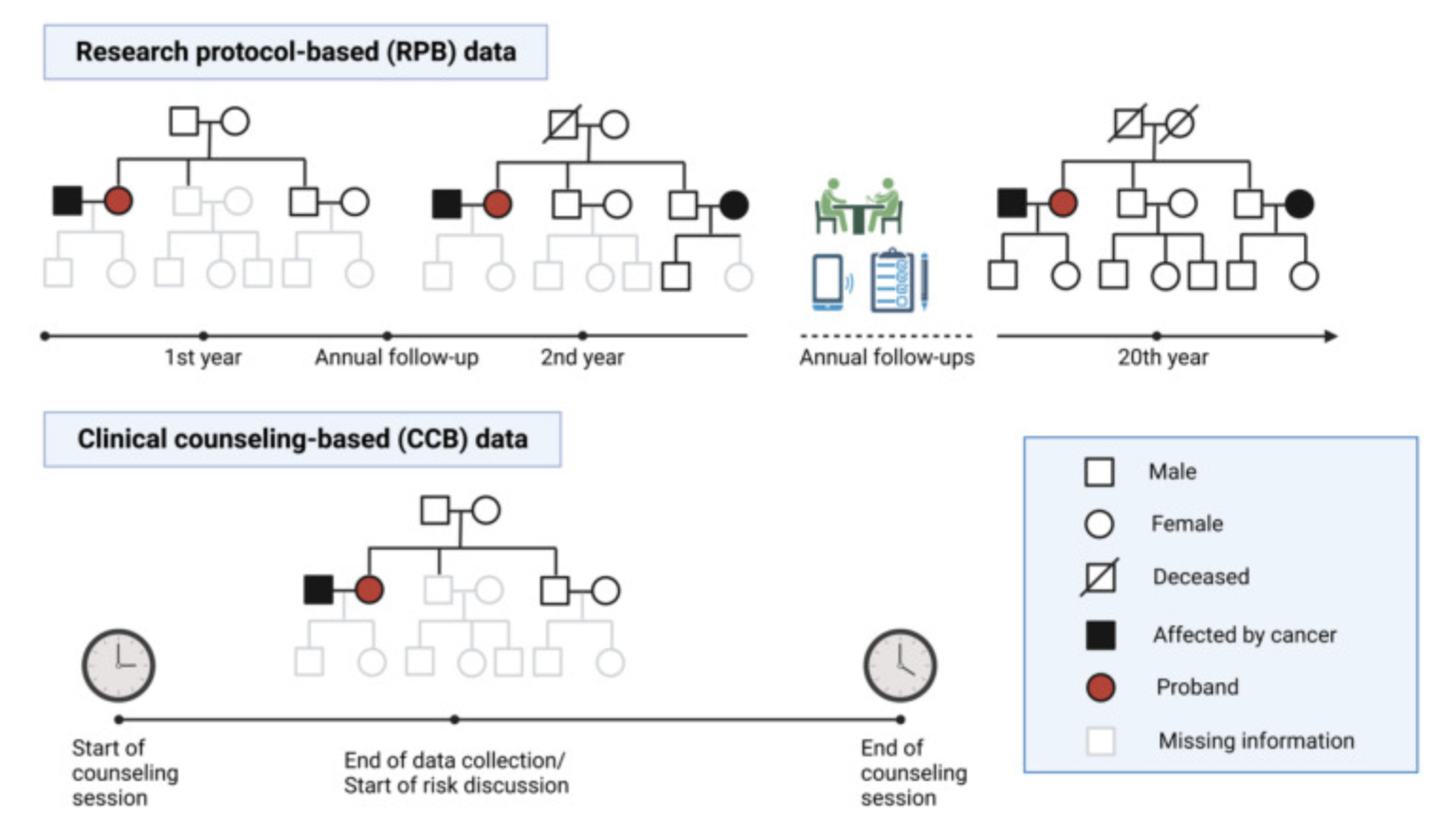
Journal of Clinical Oncology paper is online!
We conducted a validation study of our LFSPRO software suite, which was developed for risk predictions in families with Li-Fraumeni syndrome, on a clinical patient cohort collected as part of the Clinical Cancer Genetics program at MD Anderson Cancer Center. Unlike research datasets that are meticulously collected over decades for research purposes, our unique dataset closely resembles what genetic counselors observe in real counseling sessions. The validation results indicate that our risk prediction models have the potential to assist decision making in clinical settings, and further highlight the importance of such validation in bridging the gap between methodology research labs and clinics.
Free access at https://ascopubs.org/doi/10.1200/JCO.23.01926

STATS UP AI - A community for Statistics and Biostatistics
Professors from multiple universities initiate StatsUpAI, aiming to elevate
the role of statisticians in AI research.
This movement emphasizes empowering statisticians to lead and innovate in
addressing real-world challenges through AI research.
As one of the founding members, Wenyi's involvement highlights her
commitment to advancing statistical methodologies in the era of artificial
intelligence.
To learn more about StatsUpAI, visit their webpage at
https://statsupai.org
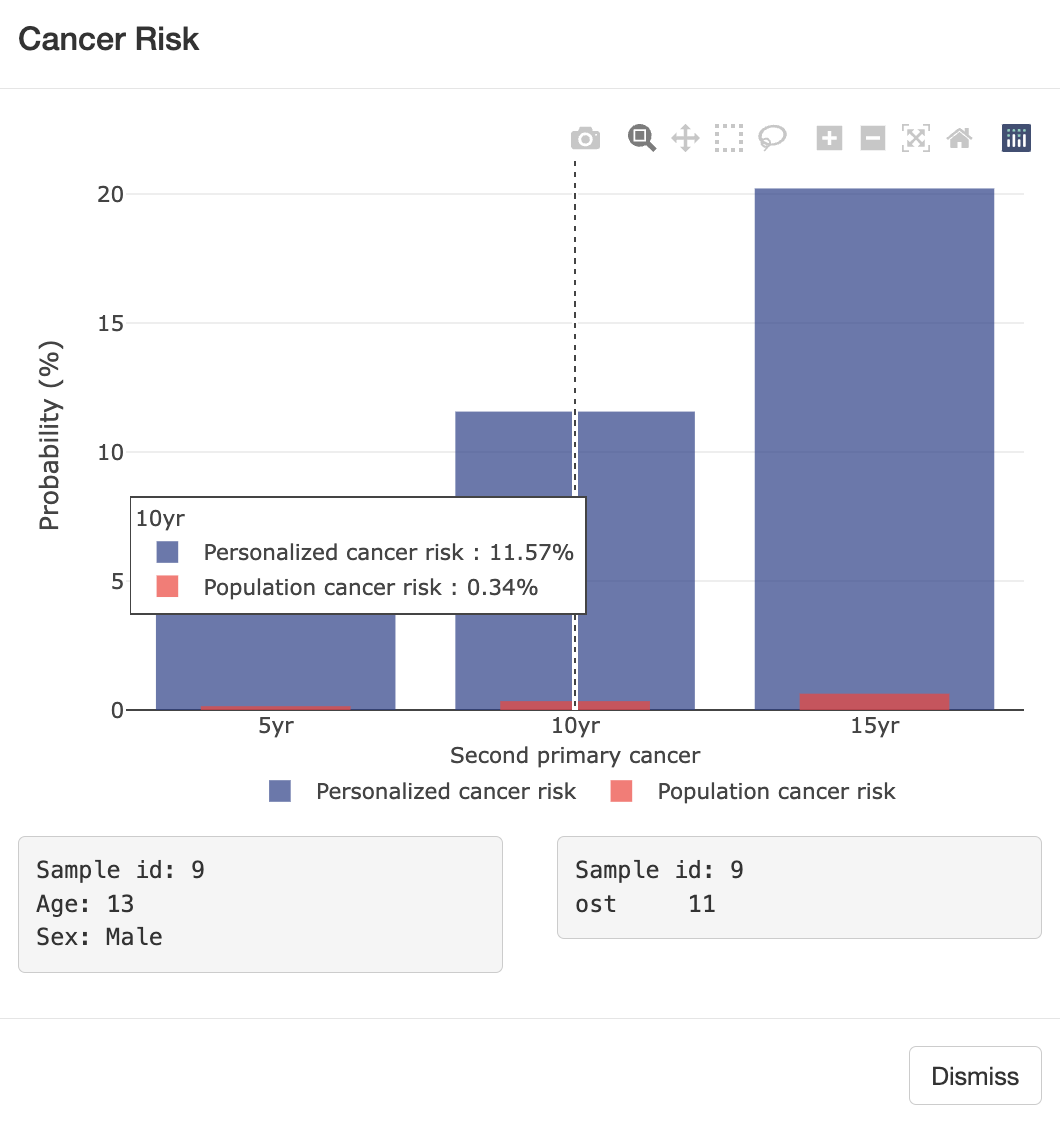
LFSPROShiny paper is published online!
LFSPROShiny is an interactive R/Shiny application designed to perform risk
prediction and visualization for Li-Fraumeni syndrome (LFS),
a genetic disorder associated with TP53 mutations, enabling genetic
counselors to assess patient risk profiles and support informed
decision-making without the need for
programming expertise.
Free access at https://ascopubs.org/doi/10.1200/CCI.23.00167.
Congratulations to the launch of Institute for Data Science in Oncology(IDSO) at MD Anderson!
IDSO integrates the most advanced computational and data science approaches
with the
institution’s extensive scientific and clinical expertise, aiming to
profoundly
enhance patient lives by revolutionizing oncological care and research.
Dr. Wang's lab is proudly affiliated with this pioneering initiative,
dedicated
to advancements in cancer care and research through innovative
methodologies.